

Markus Schilling
Markus Schilling
Technische Universität München
The Electrochemical Society hosted “Cobalt Dissolution from PtxCo/C Cathode Catalysts in PEM fuel cells: In Situ Quantification and Removal Methods,” a webinar by Markus Schilling (Technische Universität München), on April 15, 2026. A live Question and Answer session followed. Some questions not addressed during the broadcast are answered below.
Replay webinar
Q&A
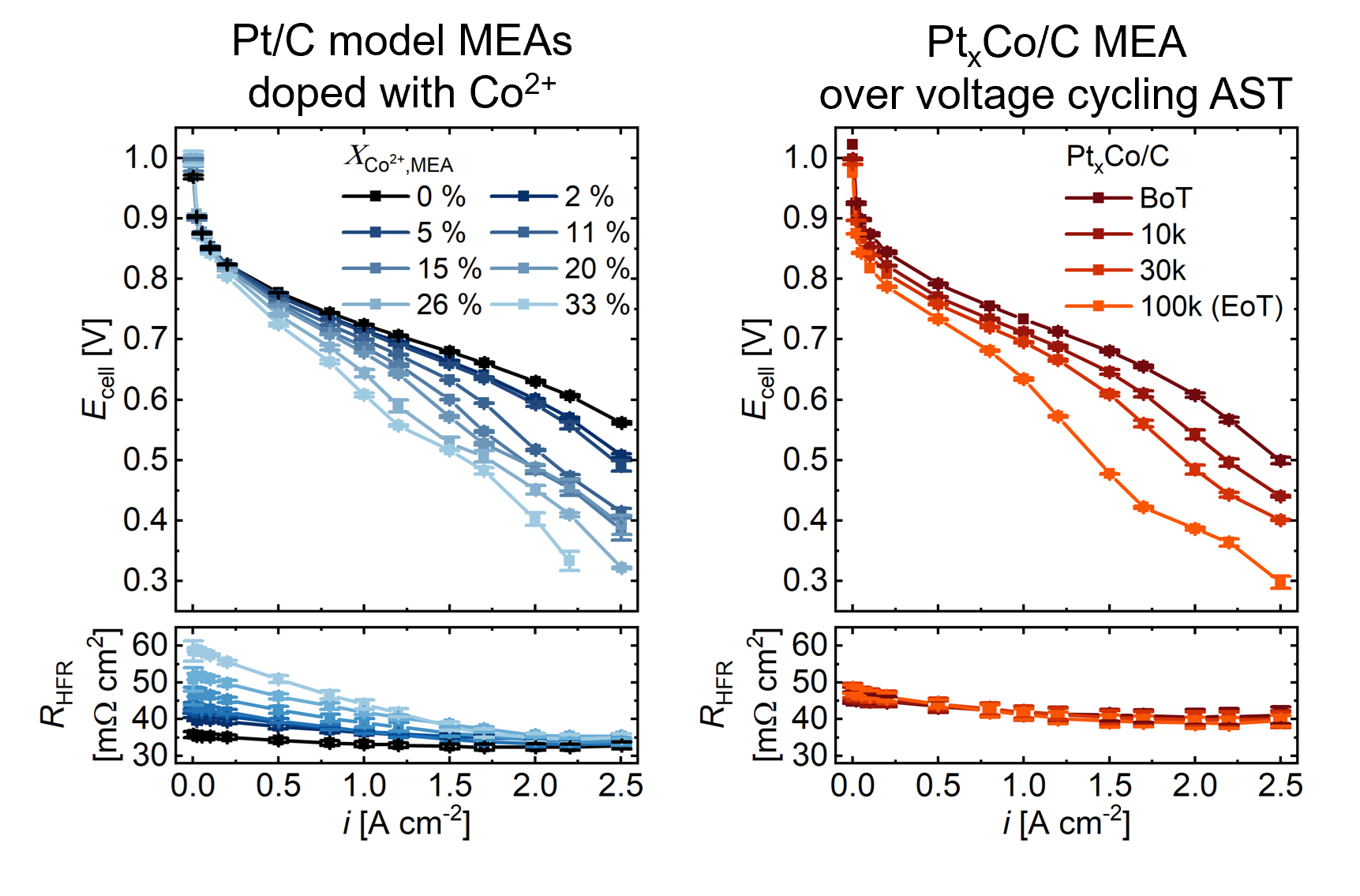
Why choose this AST protocol? Why is the AST protocol from M2FCT not recommended?
We compared various AST protocols to systematically study the impact of the upper potential limit (UPL), the hold time at the upper potential limit, and the voltage ramp rate on ECSA loss and cobalt leaching. The baseline AST protocol that we use (0.6-0.95 V, 1s-1s, square wave, H2/N2, 80 °C/95% RH) is similar to the M2FCT Catalyst AST protocol (0.6-0.95 V, 3s‑3s, square wave, H2/N2, 80 °C/100% RH). The 3s-3s hold times in the M2FCT protocol result in faster degradation per voltage cycle; however, they yield slower degradation over the total testing time than the 1s-1s hold times.
Are you considering no Co3+ when the reduction potential is near 0.28 V and could undergo redox reaction?
Based on available thermodynamic data, e.g., in Pourbaix diagrams by Chivot et al. (DOI: 10.1016/j.corsci.2007.07.002), Co2+ is the only stable species at pH≈0 at potentials that are relevant in the fuel cell (0-1 V vs RHE). However, when Co2+ ions accumulate at the cathode electrode at high current densities, the pH locally rises as protons are depleted. When the local pH in the cathode approaches seven, Co2+ ions may be oxidized and/or precipitate as Co(OH)2 or as Co3O4, depending on the potential. However, we have no direct experimental evidence for cobalt oxidation and/or precipitation yet. Once the cell current density is set to zero, the through-plane concentration profile relaxes, and Co2+ is again the only stable species at pH≈0.
For HFR measurement, what was the reason for selecting E=0.2 V and not in the double layer capacitance region like 0.45V?
The cell potential does not affect the HFR measurement, as long as the DC current is zero (i.e., in the absence of a Co2+ through-plane concentration profile). In our paper (DOI: 10.1149/1945-7111/ada188), we compare HFR measurements at 0.2 V under H2/N2 and at OCV under H2/O2, yielding virtually identical results (as for H2/air at OCV, not shown in the paper). In a stack, it might be more convenient to measure under H2/air at OCV than under H2/N2 at 0.2 or 0.45 V.
Co2+ cannot leave the ionomer phase? Does it mean Co2+ precipitates and leaves the system, which in turn explains the recovered performance? When Co is removed from Pt-Co, as observed by the HFR method, does this mean the leaching of Co improves fuel cell performance leaving behind Pt-C?
Under normal fuel cell operation (H2/air), Co2+ ions cannot leave the ionomer phase of the MEA, because there are no negatively charged counterions (except OH– at ~10-7 M). In a cation recovery with CO2:O2 mixtures as the cathode feed (DOI: 10.1149/1945-7111/ae4387), dissolved Co2+ ions can leave the MEA through the cathode exhaust water, paired with HCO3– ions, thereby preserving charge neutrality. We do not expect any cobalt precipitation under recovery conditions with CO2 in the cathode feed, because the pH of the carbonated water is acidic (pH≈4). The removal of Co2+ ions from the MEA yields a significant improvement in the H2/air performance. After removing the negative effects of Co2+ ion contamination with the CO2 recovery protocol, a PtxCo/C MEA outperforms a comparable Pt/C MEA at all current densities due to its intrinsically faster ORR kinetics.
Does the introduction of CO2 gas on the cathode side of the fuel cell have potential disadvantages that need to be considered like formation of CO gas as a poisoning agent?
Indeed, we observed a significant CO coverage (~55%) on the anode catalyst after recovery with CO2 gas in the cathode feed (see Appendix Figure A2 in DOI: 10.1149/1945-7111/ae4387). CO2 gas crossover from cathode to anode can react with adsorbed hydrogen on the anode catalyst to form adsorbed CO via the reverse water gas shift reaction. CO2 gas in the anode of a PEM fuel cell is a well-known contaminant (see, for example, Smolinka et al. DOI: 10.1016/j.electacta.2005.02.082). When the CO2 feed is stopped after a recovery, adsorbed CO at the anode can be oxidized by an anode CV or anode air bleed (in a stack). Also, standard operation under H2/air seems sufficient to oxidize most of the CO adsorbed at the anode via oxygen gas crossover from the cathode to the anode. Therefore, any losses due to CO2 forming CO at the anode are fully reversible, as shown in Figure A3 in our paper (DOI: 10.1149/1945-7111/ae4387) for a Pt/C MEA without cation contamination.
Do the carbonate ions tend to clog up or deposit on the membrane while purging CO2 in cathode during recovery?
We do not observe any precipitate after a cation recovery with CO2 in the cell or in the cathode exhaust water. Although carbonate salts of transition metal ions, such as CoCO3, have a low solubility, the CO32- concentration in the liquid water phase is negligibly small at pH≈4 (as can be calculated based on the CO2 partial pressure used during the recovery). In the Appendix Section A2 in our paper (DOI: 10.1149/1945-7111/ae4387), there is a discussion of possible precipitates, including the respective solubility products. There, we conclude that likely no precipitates form under these conditions.
Learn more about upcoming ECS Webinars and review previous webinar recordings.
Interested in presenting in the ECS Webinar Series? Email your presentation title and abstract to education@electrochem.org for consideration.
Interested in presenting in the ECS Webinar Series? Email your presentation title and abstract to education@electrochem.org for consideration.